

by Sophia Swartz
figures by Shreya Mantri
The first reports of a mysterious, pneumonia-like illness surfaced in early December 2019. Fast-forward to 2021, and the culprit—SARS-CoV-2, a virus a thousand times smaller than a speck of dust—has sickened more than 111 million people, infected all seven continents, and killed approximately 2.5 million.
The toll of COVID-19 is heart-wrenching and borders on dystopian. Our pandemic present is surreal: How could a virus that circulated in bats for millions of years make the leap into humans and suddenly plunge our world into a prolonged pandemic? And how can this virus keep changing, shapeshifting from one variant to another over several short months?
Retracing the elusive origins of SARS-CoV-2, the virus which causes COVID-19, is a scientific detective story with the power to teach us how to stay one step ahead of the virus, maintain effective diagnostic tests and vaccines, and escape over a year of quarantines and lockdowns. Surprisingly enough, this is a story that begins with a 12-letter typo.
Lean, mean, RNA machine
SARS-CoV-2 (also known as nCov-2019) is a coronavirus, a type of virus named for the distinctive “crown” of spikes coating its surface. The structure of SARS-CoV-2 is remarkably streamlined. At the most basic level, the virus can be described as a packet of RNA nucleotides (A, C, G, and U) coated with spikes.
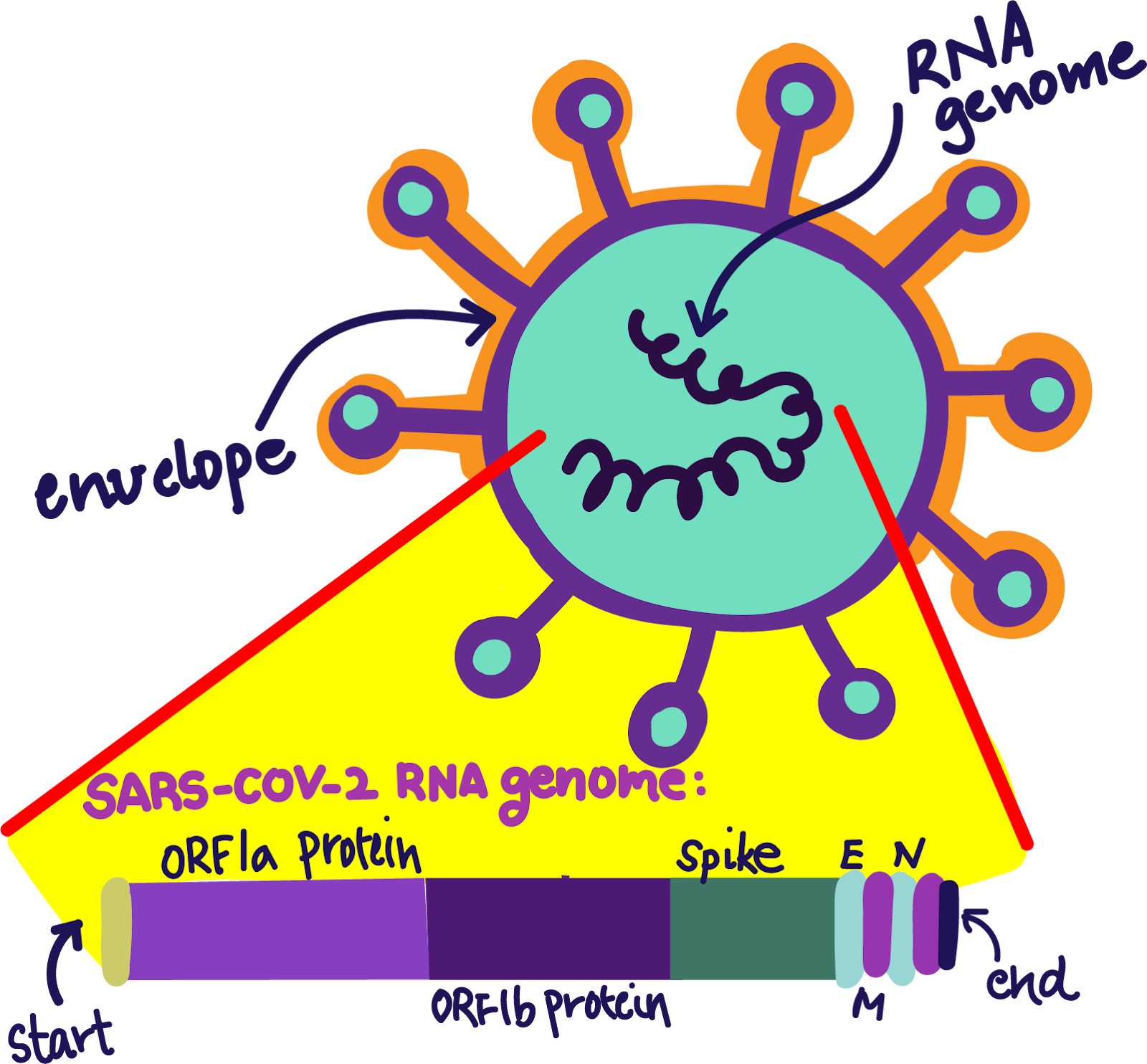
These spikes play an essential role in the spread of COVID-19. Like all viruses, SARS-CoV-2 lacks the tools to replicate, or make more copies of itself. To replicate, SARS-CoV-2 must enter and infect host cells, hijack the host cell’s machinery, and madly pump out new SARS-CoV-2 viral particles. However, entering a human host cell is not trivial. On Earth, there are more than a quadrillion quadrillion individual viruses. Only a very small fraction—about 200 viruses—can infect humans. To any average virus, uninfected human cells are like a locked titanium door; without the proper spike or “key,” it is virtually impossible for the virus to break in. And until recently, SARS-CoV-2 did not have the right key.
But at some point in late 2019, a mutation popped up in the SARS-CoV-2 genome that provided the right key. This mutation in the SARS-CoV-2 spike protein, in combination with various other factors such as proximity to human settlements, created the perfect storm for SARS-CoV-2 to cross the species divide.
An inclination for mutation
A genome is like a unique recipe for how to create an organism. The SARS-CoV-2 genome, for example, stores the template of approximately 30,000 nucleotides for how to make a SARS-CoV-2 viral particle. For SARS-CoV-2 to replicate, its genome must be copied nucleotide-for-nucleotide over a million times in just one host cell. Multiply a million copies by the number of cells in each host infected, and you quickly approach an astronomical number of copy-and-pasted SARS-CoV-2 genomes.
However, this copying process is not perfect, and nucleotide typos—called mutations—can be made. A mutation is typically a random event, and can involve adding new nucleotides, deleting old nucleotides, or even mixing up different sequences of nucleotides in a process called recombination. Scientists have even calculated an expected number of SARS-CoV-2 mutations based on how error-prone its replication is: roughly one mutation per 1,000 bases in a year. To put that number in context: if a given lineage of SARS-CoV-2 was transmitting from one person to the next for a full year, it is expected that the lineage’s viral genome would accumulate about 24 mutations. For comparison, the flu would have an average of about two mutations per 1,000 bases in one year.
Gradually accumulating mutations in a viral genome over time is normal and expected. In fact, the vast majority of these mutations either do not change how the virus behaves (null mutations) or negatively affect the virus’ ability to replicate or infect hosts (deleterious mutations). Most importantly, these mutations occur randomly. Viruses cannot maliciously plan their mutations or mutate to fulfill a specific goal. Only very rarely does a mutation in the viral genome help a virus replicate. And out of all the mutants produced normally during replication, it is only those that (1) continue to replicate and (2) display a set of inherited mutations distinct from the parent lineage that are considered variants.
Although the precise origin of SARS-CoV-2 is still unclear, scientists have tracked this variant’s leap from animals to humans down to an insertion of 12 extra nucleotides: CCUCGGCGGGCA. Whether through recombination with another viral sequence or random internal mutations, this sequence—dubbed the furin motif—unlocked human cells and created SARS-CoV-2, a human-compatible and highly-contagious lineage from the coronavirus family tree.
Surveillance, sequencing, & RNA sleuths
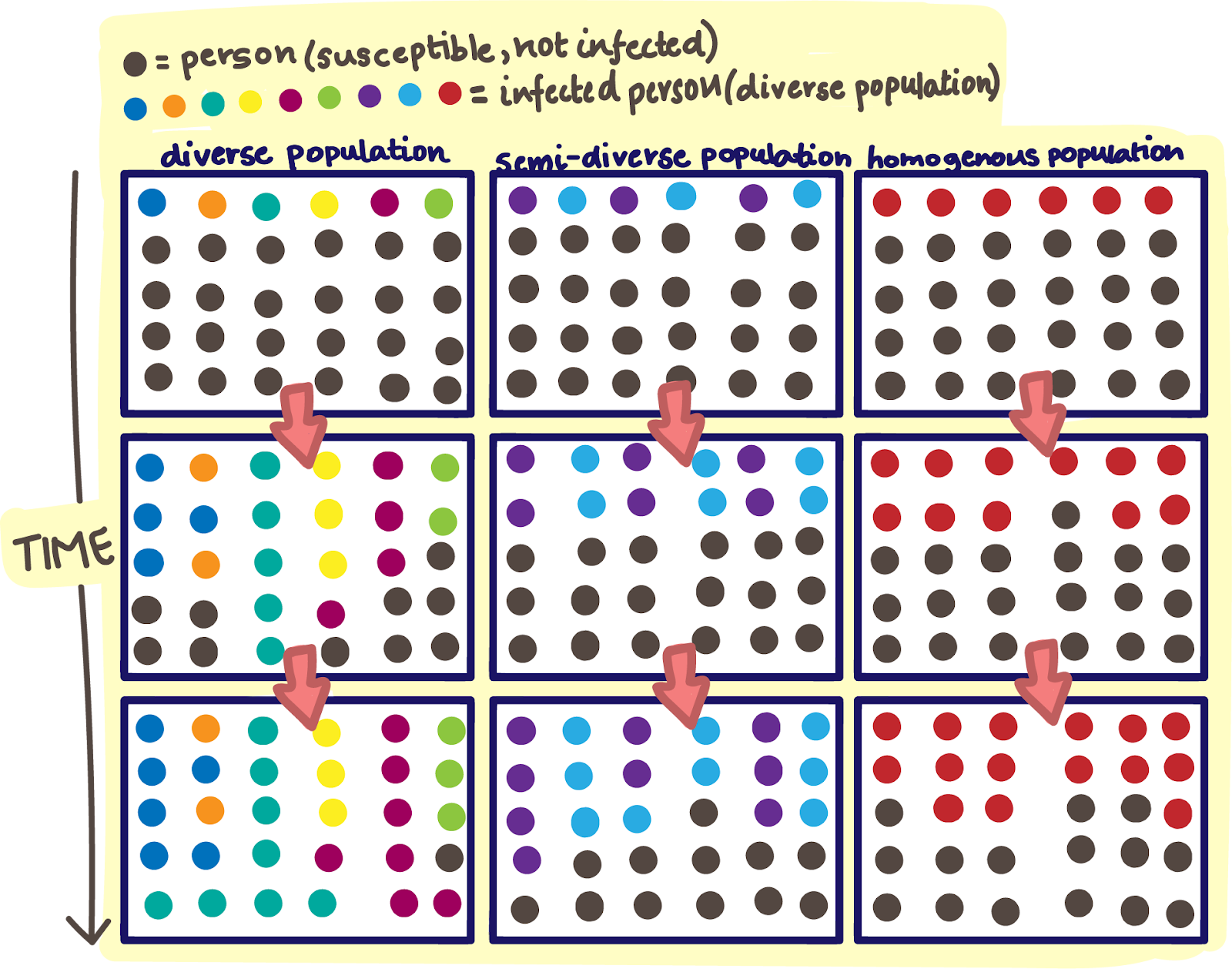
The scope and scale of a viral pandemic is hugely dependent on a variety of factors, some relating to the affected population and some relating to the virus itself. Viral factors such as virus population size and available genomic variation play key roles. Certain viruses—especially RNA viruses like SARS-CoV-2 and the flu—are better primed to create a diverse population of variants than others. SARS-CoV-2 is more resistant to genomic change than other RNA viruses, and has attained wide community spread and transmission. When roadblocks like the selective pressures of binding well to the human host cell and evading neutralization by the immune system start to stack up, variants within the SARS-CoV-2 population have an abundance of genomic back roads to rely upon.
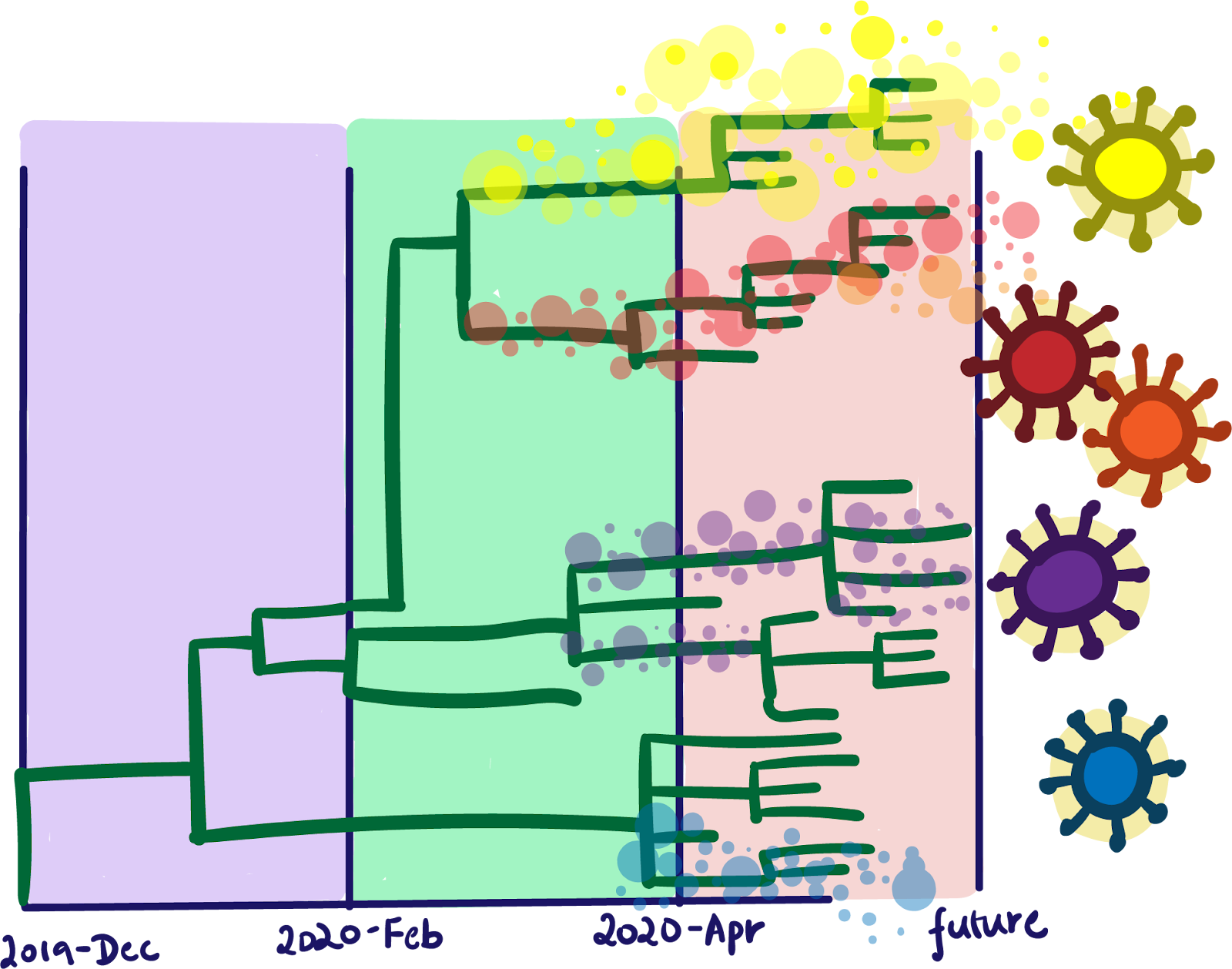
Sequencing data tracking the genomic history of SARS-CoV-2 can be immensely informative about how and when mutations arose. These data represent a complete catalog of the SARS-CoV-2 genomes sampled from an infected person, and also serve as a surveillance strategy for picking up variants and mutations silently circulating in an affected population. To-date, over 610,000 SARS-CoV-2 samples have been sequenced, allowing for powerful analyses like variant tracking and genomic epidemiology. From these samples, over 27,250 single mutations have been detected. In real-time, scientists are tracking the frequency of certain variants and mutations among COVID-19 patient samples, establishing a census of who’s who among SARS-CoV-2 variants in different countries.
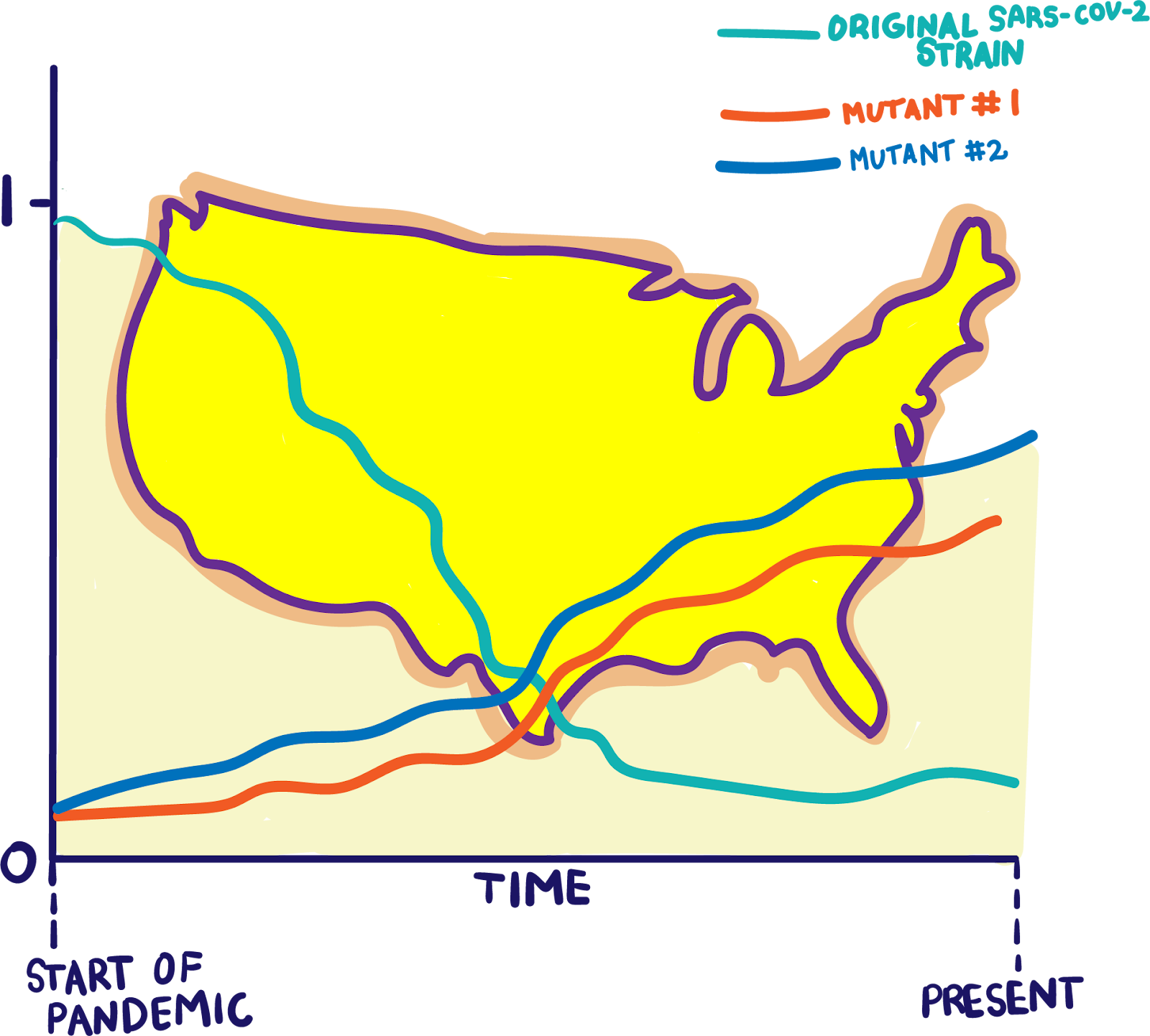
Today, the average SARS-CoV-2 genome carries 10 or fewer mutations, with most mutations acting as “passengers,” or benign genomic hitchhikers that do not outwardly affect the virus’ biology. Few SARS-CoV-2 viruses are like the B.1.1.7 variant, which sports a toolkit of 17 mutations that scientists believe may increase its infectiousness. B.1.1.7 was first detected in England in mid-September 2020. By mid-November, it represented 20 to 30 percent of COVID-19 cases in London. By early December, the proportion of COVID-19 cases caused by the B.1.1.7 variant had ballooned to more than 60 percent. Over the course of several months, the B.1.1.7 variant had outpaced its parent virus to cause a fresh surge in new COVID-19 cases in the UK. Scientists are already predicting that this pattern will happen again in other places: According to modeling by the CDC in January, the B.1.1.7 variant was projected to become the dominant SARS-CoV-2 variant in the US by April 2021. Their model is now reality: As of April 7, 2021, most US infections are caused by the B.1.1.7 variant.
Luckily, at the moment, the B.1.1.7 variant and other detected variants do not appear to escape the vaccines recently developed by Pfizer and Moderna. However, just as frequent mutations for the flu require new booster shots every year, it is entirely possible for a new SARS-CoV-2 mutation to develop that evades our current front line of defense.
If an infinite number of monkeys catch COVID-19
In 1913, French mathematician Émile Borel coined an analogy to describe the effect of different timescales on events most people instinctively consider as impossible: “If an infinite number of monkeys start playing with an infinite number of typewriters, one of them will write a play of Shakespeare.”
In this analogy, Borel used monkeys as a placeholder for any agent that randomly outputs a sequence of letters on an unfathomable scale. When considering the sheer quantity of viral genomes produced in one cell of one host during SARS-CoV-2 replication, Borel’s analogy seems well-suited to describe the adverse effects of widespread COVID-19 cases.
Just as we do, viruses follow a basic evolutionary programming to propagate, propagate, propagate. There is no design to the SARS-CoV-2 mutations that develop over time, and no intent either. Each replication cycle is a genetic roll of fair dice. However, mutations become a major concern when the virus is given a virtually unlimited number of rolls to hit the jackpot. When there is uncontrolled viral spread among millions of individuals, SARS-CoV-2 has an unimaginable number of opportunities to mutate into something much nastier than it used to be.
If drastic and sustained collective efforts are not undertaken to minimize the circulation of SARS-CoV-2 in new hosts, we are effectively fast-tracking viral evolution to outcompete our newest vaccines and ultimately leave us all defenseless. Steps like social distancing, wearing masks, and frequent handwashing can go a long way in reducing the spread of COVID-19 and the likelihood for dangerous mutants to develop.
Sophia Swartz is a junior at Harvard University studying Molecular and Cellular Biology.
Shreya Mantri is a PhD student in Biological and Biomedical Sciences at Harvard Medical School.
For More Information:
- To learn more about SARS-CoV-2 variants and mutations, check out the nextstrain.org website, which offers a wealth of interactive tools. Some highlights are linked below:
- Global genomic epidemiology map contains a phylogeny of SARS-CoV-2 variants (“Phylogeny”), a mapping of different variants around the globe (“Geography”), an analysis of sequence conservation (“Diversity”), and the proportion of cases caused by certain clades (“Frequency”)
- Situation reports detailing the progression of the COVID-19 pandemic since January 2020
- Background information on coronaviruses and how to use their analytical tools
- A paper profiling Nextstrain and how it tracks the evolution of SARS-CoV-2 in real-time
- The website CoVariants.org does an exceptional job of profiling the emergence and spread of different SARS-CoV-2 variants by country in real-time.
- For more in-depth discussion of recent variants, including the B.1.1.7 variant, explore special coverage by The New York Times here.
- If you were wondering how to picture how SARS-CoV-2 variants differ, check out this NPR Goats & Soda article describing how changes to the ACE2 receptor can affect virus transmissibility.
- Recent research has attempted to preemptively figure out what variants could emerge that would be able to escape current antibody therapies (termed “antibody escape mutants,” research article linked here) and the effect of persistent SARS-CoV-2 infections in immunocompromised patients on accelerating viral evolution.
I will bookmark it for sure. Thanks to the webmaster. Have a nice day.
Dear Sophia I just read your article (late Dec) , and I totally agree. I would like you to explore this concept of mine:
“The Virus and or its mutations are and Will never be the problem” The Real problem relies on Individual Host Heath ; specifically Endothelial Health. COVID ranges from 0-100% un terms of presentation from Asymptomatic to Severe Cases
It all depends in how much an organism is dependent to the regulatory mechanisms of ACE2. The more dependent the more sever COVID Will be. That is why heathy children have a very Mild form of the Disease.
And hence the “solution” is not the Vaccine neither their newest versions or Jabs.
The Real Solution is a proACE2 society . That understands the meaning of Mass Body Index and Metabolic Syndrome , that regulares Comorbidities and eliminates Vices ,for example Tobacco.
I truly believe SARs COV2 will be the best thing ever happened to HUMAN HEALTH (not to vulnerable individuals) , we have the natural powers to Change our cardiorespiratory behaviour and become without any vaccine a better human being.
There continues to be a debate about whether or not viruses are “alive.”
It seem that the ability of a virus to “procreate, procreate, procreate” is enough of a sign to signal that, even if a virus does not have the ability to ‘reason’ as humans do, a virus has the built-in imperative to propagate, albeit in a predatory manner, by using the host’s DNA.
Humans have followed a similar pattern: ‘procreate, procreate, procreate,” and have had, according to paleontologists, many ‘mutations’ over its million-plus span, in an obvious effort of staying “alive.”