

by Sebastian Rowe
figures by Jovana Andrejevic
First conceptualized in the 1960s, the protein folding problem – how to predict a protein’s structure from its sequence – has been one of the main concerns of structural biologists worldwide. Last year Google’s DeepMind, a team of programmers studying artificial intelligence, claimed to have the solution; much in the same way they solved the board game Go in 2016 (and made Go’s best player retire). So, will this new technology make thousands of scientists studying the structure of proteins obsolete? How did DeepMind come to this solution? And most importantly, why do we even care?
The shape and structure of proteins

“The game has changed” – Science
“It will change everything” – Nature
Proteins are the tools a living cell uses to protect itself against viruses, break down food, repair itself, and even to send messages to other cells. Much like a hammer, wrench, or screwdriver, the structure of a protein determines what the protein can do. A lot of information about how a tool is used and how a tool is likely to break can be determined from its shape. The same is true for proteins – protein structures are useful in finding new medicines. This is because most medicines work by binding (or fitting into) a protein and changing how the protein works. Most medicines stop cells from using certain proteins in ways that cause damage. Famously, the first HIV medications were designed using protein structures of the HIV protease – a protein which breaks down other proteins – in the 1990s. Scientists designed drugs that would fit perfectly in the HIV protease and turn it off. Once the structure had been determined, it only took six years for a drug to be designed, tested, and approved for use in treating HIV.
Although proteins are incredibly small (the average protein has a radius of 2nm, which is 10,000 times smaller than the width of a human hair!), there are multiple technologies that allow scientists to determine the 3-dimensional shape or structure of a protein. The most common of these technologies is X-ray crystallography, which uses the way a crystallized protein interacts with X-ray light to identify how a protein is structured. Another common method is Cryogenic Electron Microscopy (Cryo-EM), which uses electrons to take 2D photos of frozen proteins at different angles and reconstruct the protein structure in 3D. However, these technologies require the proteins to be well-behaved. For example, in X-ray crystallography the proteins must be coaxed to form crystals, and this requires very pure and highly concentrated proteins. Cryo-EM can be used for some proteins that do not crystallize well, but it does not result in highly detailed structures. Due to these problems, nearly half of human proteins do not have known structures nor are predicted to share similarity to any known structure.
A grand sudoku

The protein folding problem asks, in part, how the structure of a protein can be predicted from the amino acid sequence. Amino acids are the building blocks of every protein and each protein has a unique sequence of amino acids. Scientists consider the way amino acids interact to form a structure at different levels of depth. The sequence of amino acids that make up a protein can be determined from DNA, and this sequence is a protein’s primary structure. The way a short sequence of amino acids interacts in 3D is called a secondary structure. The tertiary structure is the structure of all of the protein’s amino acids in 3D. Knowing the tertiary structure of a protein allows scientists to design better medications, since each amino acid has many different chemical properties and these properties can change depending on what other amino acids are nearby. To determine the tertiary structure, computer programs can use these chemical properties to predict small secondary structures from the amino acid sequence. Unfortunately, every additional amino acid predicted requires a calculation of its interaction with every other amino acid, and without experimentally finding the structure, we would have no way to check that a predicted structure is correct.
The time required and the lack of a quick way to check potential solutions means that solving the protein folding problem is an example of an NP-hard mathematical problem . The “NP” stands for Non-deterministic Polynomial time, which is a way of saying the time it would take for a computer to solve a problem logically is about the same as if the computer randomly guessed and checked solutions. The “hard” in NP-hard means that we do not know if there is a quick way to check that a randomly guessed solution is correct.
Sudoku is an example of an NP problem – it is easy to check if a sudoku puzzle has the numbers placed correctly, but it’s hard to solve one. These types of problems appear everywhere in our daily life – finding the best way to commute to work or how to deliver mail are also NP-hard. If someone could find a quick way to solve or check these problems, there is a 20 year old million dollar prize waiting!
The daily commute and AlphaFold
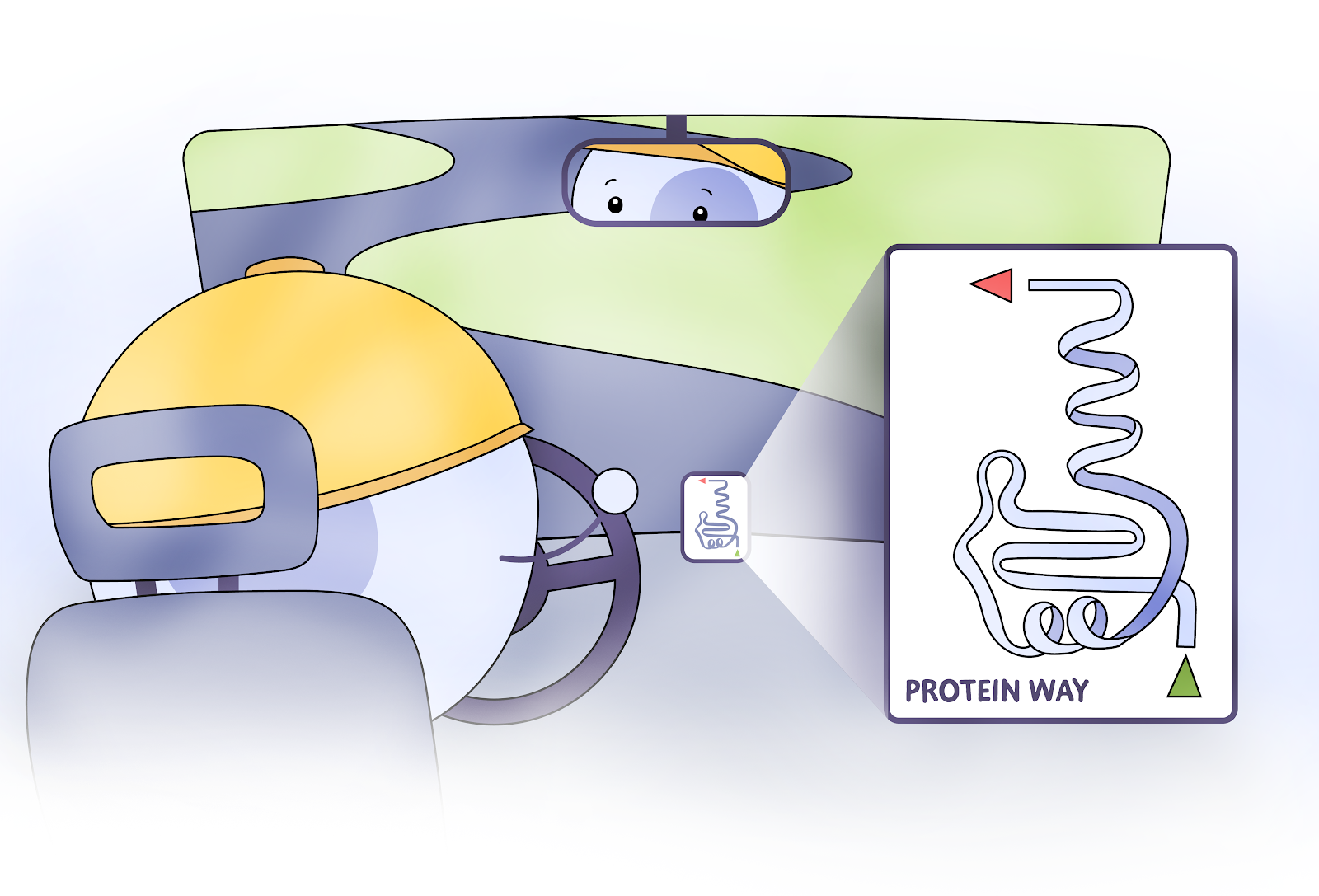
DeepMind’s AlphaFold technology is an algorithm that bypasses the need for scientists to use X-ray crystallography or cryo-EM to determine the structure of the proteins. Instead, the structure of a protein can be found by using AlphaFold on a supercomputer without involving any hands-on work. So does this AlphaFold algorithm solve the protein folding problem and will it receive the million dollar prize? Well, no. At least, it does not fully solve the problem. Instead AlphaFold has found a way to get a good enough approximation in a timeframe that is still useful rather than spend a non-deterministic (unknowably long) amount of time finding the perfect solution.
The process by which AlphaFold finds a good enough approximation is similar to how the GPS app Waze works. Waze works by deciding on a decent starting path based on thousands of previous trips and a small amount of math. Then it uses information from all the other Waze users currently on the road to update the directions as you go, accounting for small differences between the current and previous trips. In the end, the path you commute on goes from just decent to good enough – even if it is not perfect!
Similarly, AlphaFold creates hundreds of “drivers” that try different shortcuts and paths to find a tertiary structure for a particular protein with a known structure. These “drivers” are different runs of the program but starting with small random differences in the initial prediction. As these program instances calculate a predicted tertiary structure, they all share their information as they go along in an attempt to find a solution. However, it’s very possible for all the drivers to miss a possible shortcut – perhaps a roadway that isn’t on the map! For protein structures these “shortcuts” could be secondary structure motifs that are rare and not found in any known proteins. The resulting tertiary structure from AlphaFold is scored against the known tertiary structure. Then AlphaFold repeats this process for every amino acid sequence with a known structure. With every new protein structure the program trains on, the average pathfinding ability of the “drivers” gets increasingly better.
With a fully trained AlphaFold, the scientists at DeepMind entered it into the Critical Assessment of Structure Prediction – a global competition between structural biologists to predict the tertiary structures of proteins that have not been released publicly. AlphaFold was able to predict new protein structures from sequences with an error rate that averaged out to one slightly misplaced amino acid for every hundred amino acids. This error rate was the lowest rate in the competition. When using a protein structure to design new medications, scientists have to consider that there will be differences in amino acid sequences between people. As such, AlphaFold having one misplaced amino acid is within the error expected when looking at differences between the same protein from different people. This means AlphaFold is a great working solution to the protein folding problem!
A great tool for problems new and old
AlphaFold needs at least three days to find a protein structure – this is potentially years quicker than the previous methods light X-ray crystallography or cryo-EM would allow. This technology will help scientists discover new properties of proteins that have historically been hard to work with. Many of the proteins which the cell uses to communicate and interact with its environment have structures that are hard to determine. These proteins are known to be important for viruses like the one that causes COVID-19, but also for many cancers and sensory diseases. AlphaFold structures may help scientists design new medications to target these proteins. Additionally, we can use tools like AlphaFold to help us design new proteins that we can use to better deliver medications, make new kinds of materials, and make better vaccines.
Sebastian Rowe is a second-year Ph.D. student in the Chemical Biology Program at Harvard. You can find him on twitter @RuralPhD
Jovana Andrejevic is a fifth-year Applied Physics Ph.D. student in the School of Engineering and Applied Sciences at Harvard University.
For More Information:
- Dive into the press release from Google’s DeepMind on the new AlphaFold.
- Brush up on the history of structural biology with this in-depth and comprehensive overview.
- Learn more about the math behind NP problems with this great video.
- This beautiful article captures the moment DeepMind’s AlphaGo beat Lee Sedol, the world’s best Go player.